Несъвършена остеогенеза при възрастни. Несъвършена остеогенеза при деца
Има няколко различни синдрома на остеогенезис имперфекта. Заболяванията се характеризират с увреждане на костите, очите, зъбите, ушите и сърдечно-съдовата система. Тяхната класификация се основава на вида на наследяване и клиничните прояви.
Несъвършена остеогенеза тип 1
Този тип е най-често срещаният. Унаследява се по автозомно-доминантен начин и се характеризира с изразена интрафамилна вариабилност. Пациентът може да бъде нисък, да има чести фрактури и изразено намаление на работоспособността, докато негов близък роднина със същото заболяване може да живее пълноценен живот. Причината за този синдром може да са дефекти както в алфа 1(0), така и в алфа 2(1) проколаген. Мутациите често се проявяват като сини склери и нисък ръст.
Несъвършена остеогенеза тип 2
Тип 2 съчетава класическите вродени варианти, при които почти всички пациенти умират в детството или вътреутробно). Много случаи са резултат от нова мутация (доминиращ трансмисивен фенотип, ако пациентът оцелее и е фертилен) в проколаген алфа 1(1) или алфа 2(1). "Доминантно отрицателният" модел обяснява тежкия фенотип в резултат на хетерозиготна мутация. Понякога братята и сестрите на пациентите имат същите симптоми при здрави родители. В някои случаи са открити мутации в гонадите в локуси с ниски нива на експресия, което е риск от раждане на няколко болни деца.
Несъвършена остеогенеза тип 3
Тип 3 се проявява с тежка скелетна деформация, кифосколиоза, нисък ръст и чести фрактури на различна локализация. Обикновено се развива спорадично, което означава появата на нови мутации или автозомно-рецесивен тип на наследяване.
Несъвършена остеогенеза тип 4
Тип 4 е фенотипно и генетично подобен на тип 1, среща се по-рядко, не се проявява със синя склера и е свързан с по-малко фрактури.
Симптоми
Медицинската история за фрактури обикновено е подобна. „Чупките кости” е универсално проявление. Понякога фрактурите възникват вътреутробно, особено при тип 2, което прави възможна антенаталната рентгенографска диагностика. В такива случаи при раждането крайниците са къси и извити, множеството фрактури на ребрата дават вид на „броеница” на рентгенография. Пациентите с тип 1 или 4 обикновено имат анамнеза за няколко фрактури, въпреки че синята склера, опалесциращият зъбен емайл или загубата на слуха показват наличието на мутантния ген. Чупливостта и деформируемостта са резултат от дефект в колагеновата матрица на костите. Следователно скелетните прояви на остеогенезис имперфекта са наследствена форма на остеопороза. При пациенти в напреднала възраст с промени, свързани с възрастта или след менопауза, или при млади пациенти с продължително обездвижване след фрактури или ортопедични операции, рибени кости (вдлъбнатини и язви по гладките горни и долни ръбове на прешлените поради натиска на опън междупрешленния диск) или често се отбелязват плоски прешлени.
Честотата на фрактурите намалява по време на пубертета при пациенти с тип 1, 3 и 4 заболяване. Понякога след фрактура се образуват фалшиви стави. Също така доста често пациентите развиват хипертрофичен калус, от който е трудно да се различи. Въпросът за повишен риск от остеосаркома на фона на остеогенезис имперфекта остава спорен, но рискът все още е нисък, но ако възникне болка при липса на фрактура, особено при пациенти в напреднала възраст, винаги е необходимо да се изключи остеосаркома. Релаксацията на лигаментния апарат на ставите е най-силно изразена при тип 1 имперфектна остеогенеза. Луксациите са резултат от деформация, дължаща се на повтарящи се фрактури, отпускане на връзки или разкъсване на сухожилия.
Диагностика
Диференциалната диагноза на остеогенезис имперфекта включва идиопатична ювенилна остеопороза, синдром на Hajdu-Chinei (остеопороза, множество интеркаларни кости на черепа, акроостеолиза), пикнодизостоза (ръст на джудже, крехки кости, липса на мандибуларна рама, цепнати фонтанели, акроостеолиза) и хипофосфатазия. В едно от семействата склонността към остеопороза се дължи на мутация в колаген тип I. Това подчертава факта, че откриването на мутации не винаги улеснява клиничната диагноза. Освен това, прояви, които не са свързани с никакви синдроми, могат да бъдат резултат от дефекти в един или повече компоненти на извънклетъчния матрикс.
Лечение
Понастоящем са предложени няколко хормонални и фармакологични подхода за лечение на остеогенезис имперфекта. Предписването на добавки с калций, калцитонин и витамин D преди да се развие явен дефицит е неефективно. При млади пациенти, за да се намали честотата на фрактурите и да се подобри растежа на костите на скелета, приложението на бифосфонати перорално или чрез инжектиране е ефективно. Състоянието на костната тъкан като цяло не може да се подобри и все още няма препоръки относно продължителността на лечението при деца и необходимостта от лечение при възрастни. Трансплантацията на костен мозък за осигуряване на нормални мезенхимни стволови клетки е обещаваща. Генната терапия за автоложни мезенхимни стволови клетки в момента се проучва активно.
Статията е изготвена и редактирана от: хирург2959 0
Болестите на костите и ставите могат да засегнат дори медицински специалисти, които имат богат опит в своята област и са достигнали определено ниво на умения.
Но дори опитни лекари са ужасени, когато видят новородено „кристално“ лице.
Концепция и статистика
Остеогенезис имперфекта или „болест на кристалния човек” е сериозно нарушение на вътреутробното развитие на костите и ставите.
Заболяването се характеризира с повишена чупливост на костите и ставите на човешкия скелет. Обяснение за представеното заболяване може да се намери в липсата на колаген в тялото на пациента или несъответствие с нормите на описания важен протеин в структурата на костния скелет.
Болестта е генетична проява и се среща при деца, чиито родители също страдат от патологията.
В редки случаи заболяването се диагностицира при деца на напълно здрави родители и роднини. Такива характеристики се обясняват със спонтанна мутация.
Статистиката на заболяването може да доведе бременните жени до състояние на паника, тъй като процентът на фиксираните случаи е 1 новородено дете на 15 хиляди от всички бременности.
Не трябва да се поддавате на емоциите, тъй като съвременните медицински изследвания и методи на лечение могат да доведат до положителни резултати при възстановяването на болно дете.
Причини за заболяването
Както вече беше споменато по-горе, описаното заболяване е следствие от наследствена мутация на гените на колагена, което води до нарушаване на неговата структура или липсата му в организма.
Също така, проявата на „болестта на кристалния човек“ се влияе от липсата на синтезиран колаген. 
Този вид заболяване протича в по-лека форма и се характеризира с повишена степен на чупливост на костите, което води до чести фрактури при болния. След пубертета броят на счупванията значително намалява, а в зряла възраст всичко се повтаря.
Експертите не могат да обяснят причините за спонтанната мутация. Единственото нещо, което може да се посъветва на бременните жени, е да бъдат внимателни към здравето си и да се подлагат на редовни прегледи по време на бременност.
Не пийте алкохол и спрете да пушите, докато човек е в утробата.
Видове и симптоми на заболяването
Osteogenesis imperfecta има няколко форми на проявление и развитие, които се характеризират с отличителни симптоми и структура на костите на болния човек.
Тип I – слаба форма
Броят на хората, страдащи от този тип, е приблизително 50% от всички идентифицирани случаи. Както вече споменахме, пациентите са податливи на чести фрактури на костите и дислокации на ставите.
Рискът от фрактури намалява след 10 години живот, но след 40 години пациентът се връща в рисковата група.
При първия тип настъпват определени промени в аортата, в резултат на което могат да се наблюдават чести кръвотечения от носа.
Тип II – перинатално-летален
Тази форма на проявление на остеогенезис имперфекта се характеризира с честа смърт на плода по време на бременността на жената. В противен случай преждевременното раждане се случва в кратки срокове на бременността. Тук също има три групи:
- Група А– наранявания на главата се записват дори на етапа на вътрематочно развитие. Децата се раждат с ръст само 20-30 см. Нарушенията на мозъчната дейност и дихателната система са ясно изразени. Новородените са или мъртвородени, или умират през първите няколко дни (в редки случаи умират в края на първия месец от живота). Смъртта на детето е причинена от множество фрактури.
- Група Б– признаците на заболяването са същите като при група А, с изключение на нормалното развитие на дихателната система или с леки отклонения от нормата. Такива новородени могат да живеят няколко години. Имат скъсяване на всички тръбести кости.
- Група Б- Диагностицира се много рядко. Новородените умират в първите дни от живота си или вече се раждат мъртви. Отбелязват се изтъняване на тръбните кости и липса на осификация на черепа.
Тип III - нарушение на растежа на костите
Тип III е изключително рядък и се характеризира с нарушен растеж на костите.
Тялото на новородено с нисък ръст може да има нормално тегло. Също така се диагностицират нарушения на кръвообращението, което в повечето случаи причинява смърт. Фрактурите на костите се записват по време на раждане.
Тип IV - нарушения на растежа на скелета
Тип IV се характеризира с наличието на скелетни аномалии. След няколко години пациентът развива костни калуси и броят на фрактурите намалява. След 30 се наблюдава загуба на слуха.
Цялостната диагностика, която се извършва веднага след раждането на бебето, помага да се идентифицира вида или групата на патологията.
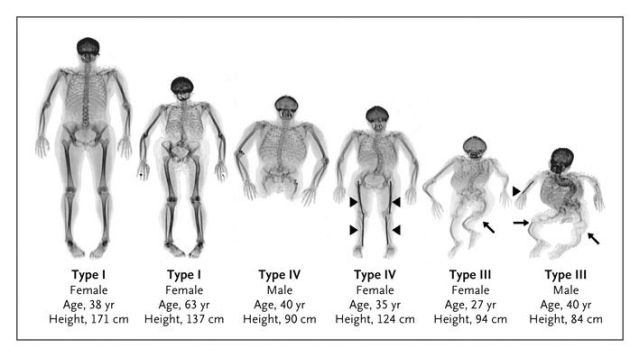
Снимката показва четири вида имперфектна остеогенеза
Диагностика на заболяването
Диагнозата се извършва на два етапа. Трябва също да се отбележи, че има вътрематочна диагноза. Тук се използва ултразвук.
Ако има съмнения за нарушения на структурата на колагена, се извършва допълнителна серия от химични изследвания с вземане на амниотична течност и епителна тъкан от бременната жена.
Веднага след раждането на болно бебе се извършват редица инструментални изследвания, които включват:
- Рентгенова снимка - чрез изображението можете да идентифицирате съществуващи фрактури;
- денситометрия - извършва се изследване на минералната кухина на костната тъкан;
- Костната тъкан се събира за биопсия.
В допълнение към инструменталните изследвания се извършват и лабораторни изследвания:
- въз основа на кръвта се откриват аномалии в структурата на ДНК;
- извършват изследвания за диагностика на колаген;
- направете няколко теста въз основа на кожна биопсия.
Въз основа на диагнозата специалистите съставят подходящ план за лечение.
Лечение на патология
 Osteogenesis imperfecta се лекува с помощта на няколко метода, включително медицински метод, базиран на употребата на лекарства от пациента за увеличаване на костната плътност, за да се намали броят на фрактурите.
Osteogenesis imperfecta се лекува с помощта на няколко метода, включително медицински метод, базиран на употребата на лекарства от пациента за увеличаване на костната плътност, за да се намали броят на фрактурите.
Съставът на лекарствата трябва да включва калций, витамин D, калиеви соли, магнезий и други полезни химикали.
В допълнение към лекарствената терапия се използват физиотерапия и терапевтични упражнения. Също така лечението се основава на психотерапия, която се провежда от родителите на болното дете и други роднини.
Задачата на психотерапията е да обясни основните правила и методи за по-нататъшно обучение на детето как да се държи в обществото, така че да може самостоятелно да избягва ситуации, които могат да доведат до фрактура.
Усложнения и прогноза
Невъзможно е да предотвратите и предпазите детето си от развитието на патология, но с навременното лечение и изпълнението на всички съвети и препоръки, изразени от лекаря, новородените бебета могат да растат и да се реализират в живота, тъй като характерните психологически или умствени аномалии са не се открива при деца. 
Усложнения могат да бъдат причинени от падане, тъй като за "кристалния" човек лек удар с топка по гърба по време на игра на футбол може да доведе до фрактура на гръбначния стълб.
Ето защо животът на кристалните хора не може да се сравни с живота на здравите хора.
Те трябва да се пазят от най-малките наранявания и натъртвания; всяко падане може да доведе до счупване на крака и последващо поставяне в инвалидна количка.
Ако родителите приемат детето си и започнат подходящи действия за неговото възстановяване, могат да се постигнат добри резултати и положителна динамика.
По правило няма да последва пълно възстановяване, но благодарение на съвременния технологичен прогрес животът на пациента може значително да се улесни.
Водещи експерти в областта на генетиката:
проф. Круглов Сергей Владимирович (вляво), Крючкова Оксана Александровна (вдясно)
Редактор на страници:Крючкова Оксана Александровна - травматолог-ортопед
 Амелина Светлана Сергеевна - професор в катедрата по генетика и лабораторна генетика, доктор на медицинските науки. Лекар генетик с най-висока квалификационна категория
Амелина Светлана Сергеевна - професор в катедрата по генетика и лабораторна генетика, доктор на медицинските науки. Лекар генетик с най-висока квалификационна категория
 Дегтерева Елена Валентиновна - асистент на катедрата по генетика и лабораторна генетика, генетик първа категория
Дегтерева Елена Валентиновна - асистент на катедрата по генетика и лабораторна генетика, генетик първа категория
Остеогенезис имперфекта е генетично заболяване, характеризиращо се с факта, че на генетично ниво има нарушение на образуването на костите, в резултат на което тези кости, които се образуват при дете, имат пореста структура (наблюдава се остеопороза) и прекомерно повишена степен на крехкост.
Несъвършена остеогенеза: епидемиология

В медицинската литература концепцията за имперфектна остеогенеза има няколко различни наименования – вродена чупливост на костите, вроден рахит, периостална дистрофия, болест на Фролик-Лобщайн и вродена остеомалация. Но въпреки такова разнообразие от имена, всички те отразяват един патологичен процес, протичащ в костните структури.
Поради прекомерната крехкост и чупливост на костите, децата с това заболяване са склонни към постоянни и най-важното - множество фрактури на костите. Освен това фрактури се получават дори от най-лекия удар, който при здрави деца изобщо не причинява травматично увреждане. Във връзка с тази особеност децата, страдащи от тази патология, понякога се наричат „кристални деца“, като по този начин се подчертава особеността на костните структури, които се наблюдават при тях.
Честотата на тази патология сред населението на света е 1:10 000 или 1:20 000 от всички новородени деца.
Osteogenesis imperfecta е генетична патология. Това означава, че е невъзможно да се излекуват напълно пациенти, на които е поставена тази диагноза при раждането, както при всички други деца с генетични проблеми. Но въпреки това вече са разработени редица методи, които спомагат за нормализиране на функционирането на такива пациенти, повишавайки качеството им на живот.
Несъвършена остеогенеза: причини за развитието на тази патология.
Така че, нека да разберем защо възниква тази патология, какви механизми в тялото на детето са „повредени“.
Развитието на тази патология се дължи на факта, че на генетично ниво (поради мутация) има нарушение на метаболизма на колагенови протеини тип 1 (протеини на съединителната тъкан). Поради такова нарушение се нарушава образуването на колагенови вериги. В резултат на това образуването на колагенови влакна от вериги, които изграждат както костите, мускулите, така и всички други съединителни тъкани, е напълно или частично нарушено. И именно това нарушение вече води до факта, че костните структури се образуват с дефект. Как точно се изразява това нарушение? Изглежда, че костта расте както обикновено по отношение на дължината си, но въпреки това се осифицира слабо (нарушени са периосталните и ендосталните видове осификация). Структурата на костта става пореста - отделни костни островчета и многобройни синуси, които са изпълнени с рехава съединителна тъкан. Кортикалния слой, покриващ костта, е по-тънък от нормалното. В резултат на всички изброени по-горе промени, дори и при най-лекия удар се образуват многобройни фрактури, които по принцип не би трябвало да се случват.
Унаследяването на тази патология може да възникне или чрез автозомно-доминантен тип наследяване (до деветдесет и пет процента от всички случаи на тази патология) или чрез автозомно-рецесивен тип (този тип наследяване представлява по-малко от пет процента от всички регистрирани случаи на болестта). Също така, проявата на това заболяване в приблизително петдесет процента от всички случаи е спонтанна мутация.
Несъвършена остеогенеза: класификация
1) Тип 1 на остеогенезис имперфекта: унаследява се по автозомно-доминантен начин. Според степента на протичане е лека до средно тежка. Този тип се характеризира с наличие на фрактури, но тежестта на фрактурите е умерена и възниква остеопороза. В допълнение към тези прояви се отбелязват следните:
· Синьо оцветяване на склерата
Развитие на ранна загуба на слуха при дете
· Нарушено развитие на зъбите
Ако пациентът има всички тези признаци, тогава това е подтип 1А. ако пациентът няма проблеми със зъбите, тогава това е подтип 1В.
2) тип 2 имперфектна остеогенеза. Унаследява се по автозомно-рецесивен начин. Според тежестта на протичане протича под формата на тежка перинатално-летална форма. В клиничната картина се наблюдава липса на осификация на черепа, намаляване на обема на гръдния кош, промени в ребрата (приемат формата на броеница), деформация на дългите тръбести кости. Появата на фрактури при този тип се случва в пренаталния период.
3) тип 3 имперфектна остеогенеза. Унаследява се по автозомно-рецесивен начин. Клиничната картина включва:
o костна деформация. Има прогресивен характер
o Dentinogenesis imperfecta
o Развитие на фрактури. Освен това те се появяват през първата година от живота на детето.
4) тип 4 имперфектна остеогенеза. Типът на наследяване е автозомно доминантен. В клиничната картина:
Ø Малка височина
Ø Деформация на скелета
Ø Имперфектна дентиногенеза
Ø Оцветяването на склерата е нормално
Ø Счупвания на кости
Несъвършена остеогенеза: етапи на развитие на заболяването
§ Латентен стадий
§ Стадий на патологични фрактури
§ Стадий на глухота
§ Стадий на остеопороза
Възможно е да се комбинират прояви на остеогенезис имперфекта с други наследствени заболявания, като микроцефалия, катаракта и вродени контрактури на ставите.
Osteogenesis imperfecta: симптоми на заболяването
Клиничните прояви и тежестта на заболяването зависят от генетичния тип.
v Вътрематочна форма. Децата се раждат мъртвородени. Ако дете се роди живо, то умира през първите седмици - първия месец след раждането си (до осемдесет процента от всички случаи). Освен това:
I. Интракраниални наранявания, получени вътреутробно или при раждане
II. Синдром на респираторен дистрес
III. Инфекции, често засягащи дихателната система
IV Кожата на такива пациенти е тънка и бледа
V. Практически няма подкожна мастна тъкан
VI. Хипотония
VII. Множество фрактури (бедрена кост, подбедрица, предмишница, рамо, рядко - ключица, гръдна кост, тела на прешлени)
v Късна форма на остеогенезис имперфекта.
Типична триада от симптоми:
а. Повишена чупливост на костите, главно в долните крайници
b. Оцветяване на склерата - синьо
c. Прогресивна загуба на слуха, водеща до глухота.
Освен това се отбелязва:
аз Фонтанелите се затварят твърде късно в сравнение със здравите деца
ii. Детето изостава във физическото развитие
iii. Ставите са разхлабени
iv. Мускулите са атрофирани
v. Има луксации/сублуксации в ставите
vi. Фрактури. Те се появяват дори при опит за повиване на дете, къпане или обличане.
vii. Деформацията на крайника, неговото скъсяване се развива поради наличието на фрактури и тяхното неправилно сливане.
viii. Деформация на гръдния кош
ix. Изкривяване на гръбначния стълб
Ако детето има дентиногенезис имперфекта:
Зъбите пробиват много по-късно от очакваното (след две години), захапката е с необичайна форма, зъбите са жълти на цвят („кехлибарени зъби“), изтъняват и се износват твърде бързо, много лесно се разрушават и са податливи на множество кариеси .
Несъвършената остеогенеза може да се комбинира с редица заболявания като напр
1. Пролапс на митралната клапа
2. Митрална недостатъчност
3. Изпотяване. Свръхекспресиран
4. Уролитиаза (бъбречни камъни)
5. Херния (ингвинална, пъпна)
6. Кървене. Основно назално
Несъвършена остеогенеза: диагностични мерки
1. Пренатална диагностика. Открива наличието на патология дори на етапа на бременността. Акушерско ултразвуково изследване се извършва след шестнадесетата седмица от бременността. Ако е необходимо, биопсия на хорион и ДНК диагностични изследвания се извършват стриктно по предписание на генетика.
2. Рентгеново изследване на кости и стави. Рентгеновото изследване може да разкрие наличието на множество фрактури, костни деформации, остеопоротични промени и намаляване на дебелината на кортикалния слой.
3. Хистоморфометрично изследване. Извършва се по време на пункционна биопсия на илиачното крило и кожна биопсия.
4. Генетични тестове
5. Консултации със специалисти (генетик, ортопед-травматолог, педиатър, зъболекар, УНГ).
Несъвършена остеогенеза: лечение
1. Костната минерална плътност се нормализира. Възможно е да се използват бифосфанати, лекарства, които намаляват скоростта на разрушаване на костната тъкан.
2. Предотвратяване на счупвания, удари, травматични увреждания
3. Психическа, физическа, социална рехабилитация на пациенти
4. Лечебна гимнастика
6. Хидротерапия
7. Физиотерапевтично лечение - ултравиолетово лечение, електрофореза с калциеви соли, магнитотерапия.
8. Витамини от група D
9. Мултивитамини
10. Лекарства, съдържащи фосфорни и калциеви соли.
11. Соматотропин. Предписва се за стимулиране образуването на колагенови влакна. След приключване на курса на лечение с това лекарство се използват лекарства, които ускоряват минерализацията на костите.
12. Ако има фрактури, поставете гипсови отливки след сравняване на фрактурата.
13. При наличие на деформации, които са изключително изразени, се извършва оперативна интервенция за отстраняването им.
Несъвършена остеогенеза: прогноза на заболяването
Ако пациентите имат вродена форма, смъртта настъпва през първите месеци от живота на новороденото. При наличие на късна форма протичането на заболяването е благоприятно, въпреки че качеството на живот е ниско. Необходимо е да се грижите правилно за такива деца, да ограничите травматичните наранявания и да провеждате курсове на лечение и рехабилитация. Ако в семейството вече има пациент с установена диагноза остеогенезис имперфекта, тогава двойката, която планира да има дете, се препоръчва да премине генетично консултиране, последвано от изследване.
За да си запишете час при генетик:
Уважаеми пациенти, Предоставяме възможност за регистрация директнода видите лекаря, който искате да посетите за консултация. Обадете се на посочения в горната част на сайта номер, ще получите отговор на всички ваши въпроси. Първо, препоръчваме ви да проучите раздела За нас.
Как да си запишете час при лекар?
1) Обадете се на номера 8-863-322-03-16 .
2) Ще ви отговори дежурният лекар.
3) Говорете за това, което ви притеснява. Бъдете готови, че лекарят ще ви помоли да ви разкаже възможно най-подробно за вашите оплаквания, за да определи специалиста, необходим за консултация. Дръжте под ръка всички налични тестове, особено тези, направени наскоро!
4) Ще се свържем с вас бъдещелекуващ лекар (професор, доктор, кандидат на медицинските науки). След това ще обсъдите мястото и датата на консултацията директно с него - с човека, който ще Ви лекува.
При този тип имперфектна остеогенеза при новородени, костната крехкост и множество фрактури са изразени, което води до прогресивна скелетна деформация. Синият цвят на склерата при новородено става по-малко забележим, когато детето расте. Заболяването се унаследява по автозомно-цесивен начин; голямото разнообразие от клинични форми показва генетична хетерогенност.
Много малко пациенти оцеляват до зряла възраст. Теглото и дължината на новороденото обикновено остават непроменени, но последното скоро намалява поради деформация на краката. Счупванията, които в повечето случаи възникват при раждането, често се случват по-късно. Кифосколиозата, която се развива в детството, прогресира при юноши. Дължината на тялото в крайна сметка става много кратка. Децата с тази форма на синдрома имат загуба на слуха. Значителна част от тях умират от кардиопулмонални усложнения.
Рентгенологично се отбелязва генерализирана остеопения: с множество костни фрактури, без ясно оформени ребра или модел на смачкани фрактури на дълги кости, характерни за тип II. Прогресията на остеопения води до развитие на платиспондилия; прешлените придобиват форма, подобна на тази на рибата треска. Черепът е мек с малки червееви островчета на осификация.
Несъвършена остеогенеза тип IV. Този тип синдром се проявява само с остеопороза, водеща до чупливост на костите, без други класически характеристики на остеогенезис имперфекта тип I и се характеризира с автозомно-доминантен начин на унаследяване. Синкавите склери на новородено стават по-светли с растежа на детето и до зряла възраст почти не се различават от нормата. Слухът не се променя. Някои членове на семейството имат опалесциращ дентин, което показва хетерогенност на заболяването.
Osteogenesis imperfecta тип IV може да се развие от раждането или по време на юношество и зряла възраст. Тежестта на деформацията на дългите кости варира в широки граници. Изкривяването на краката може да бъде единственият клиничен признак на синдрома при новородено; при някои пациенти прогресивната деформация на дългите кости не е придружена от фрактури. Изкривяването на костите намалява значително с възрастта. При някои пациенти, след достигане на пубертета, фрактурите се появяват по-рядко. Дължината на тялото на повечето пациенти е малка. Рентгенологично се отбелязва широко разпространена остеопения. Множество фрактури могат да възникнат при раждането и през целия живот. Остеопенията е по-слабо изразена и честотата на фрактурите е по-ниска, отколкото при деца с рецесивни форми на остеогенезис имперфекта.
Лечение на имперфектна остеогенеза. Няма ефективни лечения за пациенти с остеогенеза имперфекта тип II. При други видове те се състоят предимно от внимателно боравене с новородените, а използването на твърди матраци или възглавници при повиване помага за предотвратяване на големи фрактури. Впоследствие активната ортопедична тактика, състояща се от незабавна репозиция на фрагменти и обездвижване на крайниците в случай на фрактури и коригиране на последствията от прогресивното изкривяване на скелета, придобива голямо значение. Лечението с калциеви или флуорни препарати, аскорбинова киселина или магнезиев оксид е неефективно. Някои изследователи съобщават за увеличаване на костната маса и намаляване на честотата на фрактурите с калцитонин, който в момента е подложен на клинични изпитвания. При предоставяне на генетично консултиране на роднини на пациенти трябва да се препоръча преди всичко превенция на заболяването. Невъзможно е да се постави точна диагноза по време на развитието на плода, но в някои случаи изразената остеогенеза имперфекта тип II може да бъде диагностицирана с помощта на ултразвукови и рентгенови методи.
Остеопороза с псевдоглиоматозна слепота. Този рядък автозомно-рецесивен модел на унаследяване се характеризира с генерализирана остеопороза, водеща до фрактури и деформации на дългите кости и гръбначния стълб. Очен псевдоглиом, който се развива при кърмачета, често се бърка с ретинобластом. Леко умствено изоставане при някои пациенти може да не е свързано с болестта.
Osteogenesis imperfecta е генетично заболяване, свързано с нарушено костно образуване. Заболяването се проявява от раждането с патологична чупливост на костите, мускулна слабост и нарушен растеж.
Въз основа на клиничните характеристики има четири основни вида на заболяването.
Лечението на остеогенезис имперфекта е симптоматично, тъй като заболяването е генетично по природа.
Причини за имперфектна остеогенеза
Osteogenesis imperfecta се унаследява по автозомно-доминантен начин, но се срещат и автозомно-рецесивни форми на заболяването.
Причината за остеогенезис имперфекта е нарушение на минералния или протеиновия метаболизъм, повишена активност на остеокластите или намалена функция на остеобластите. При остеогенезис имперфекта настъпва качествена промяна във функционалната активност на тези клетъчни елементи. Голям брой остеобласти, които имат висока пролиферативна активност, произвеждат малко количество костна материя и бързо се трансформират в остеоцити.
Според съвременните изследвания при остеогенезис имперфекта не се произвежда достатъчно колаген - произвеждат се главно предколагенови влакна, които не са подложени на узряване, или специален качествен състав на колаген.
Видове и симптоми на имперфектна остеогенеза
Заболяването се класифицира в четири основни вида. Наскоро бяха идентифицирани и типове V, VI, VII и VIII:
- Тип I имперфектна остеогенеза. Счита се за най-леката форма на заболяването. Характеризира се с наличието на синя, синя или сиво-сива склера при дете, ранна загуба на слуха, умерени промени в костите, лека кривина на гърба, намален мускулен тонус, слабост на лигаментния апарат, леко изпъкнали очи;
- Тип II на заболяването се характеризира с такава тежка чупливост на костите, че фрактурите се появяват дори в пренаталния период, което често води до смърт на плода. Следователно този тип се нарича още перинатално-смъртоносен. Ако се роди дете, то най-често умира през първата година от живота поради вътречерепен кръвоизлив или дихателна недостатъчност;
- Тип III се характеризира с прогресивни тежки деформации; проблеми с дишането; несъвършена дентиногенеза; нисък ръст, изкривяване на гръбначния стълб; слаб мускулен тонус и връзки; ранна загуба на коса. Този тип се нарича още тип прогресивна деформация, при който новороденото проявява леки симптоми на заболяването, които се засилват с израстването на детето. Продължителността на живота на такива пациенти може да бъде нормална, но с доста сериозни пречки пред живота;
- Тип IV се характеризира с лека костна чупливост (особено преди пубертета), костна деформация, варираща от лека до умерена; изкривяване на гръбначния стълб; варел ракла; ранна загуба на коса.
Има две форми на това заболяване: вродени и късни.
Твърди се, че има вродена форма, ако детето е родено с деформации на крайниците в резултат на вътрематочни фрактури.
Късната форма на остеогенезис имперфекта се среща в по-напреднала възраст.
Типичните признаци на това заболяване са:
- Чести патологични фрактури. Най-чести са счупванията на гръбначния стълб и дългите кости. Техният брой не зависи от формата на заболяването;
- Ранна загуба на слуха. Глухотата при такива пациенти може да настъпи след навършване на 10 години;
- Очни прояви. Цветът на склерата при пациенти с остеогенезис имперфекта може да варира от нормален до леко синкав или от сиво-син до ярко син. Синият нюанс е свързан с прозрачност или изтъняване на колагеновите влакна на склерата, през които се вижда хориоидеята;
- Дефекти на дентиногенезата. Зъбите с това заболяване имат жълтеникаво-кафяв, кехлибарен или синкаво-сив полупрозрачен цвят поради неправилно отлагане на дентина. Млечните зъби като правило са по-малки от кътниците; постоянните имат основа и са като че ли заострени. В този случай зъбите пробиват късно и доста често са засегнати от кариес.
Много пациенти също проявяват аномалии на кожата и ставите, сърдечно-съдови нарушения, хипертермия и прекомерно изпотяване.
Диагностика на остеогенезис имперфекта
Диагнозата на това заболяване се основава предимно на резултатите от рентгеновото изследване.
При поставяне на диагнозата трябва да се изключи хондродистрофия, която може да се подозира поради симптома на микромелия, който е общ за двете заболявания; рахит. Това заболяване се диференцира и от нефрогенните остеопатии, при които настъпват промени във фосфорно-калциевия метаболизъм; синдром на van der Hoeve.
Лечение на имперфектна остеогенеза
При това заболяване лечението не е ефективно и се свежда главно до симптоматична терапия.

Целта на лекарствената терапия е да засили синтеза на колаген, да активира хондрогенезата и процеса на минерализация на костната тъкан. Соматотропинът се използва като стимулатор на протеиновия синтез.
В същото време се извършва електрофореза на тръбни кости с калциеви соли, магнитна и индуктотерапия, предписват се витамини С, В1 и В6, тренировъчна терапия и масаж.
Особено внимание се обръща на ортопедичното и хирургично лечение на остеогенезис имперфекта. Операцията се използва за коригиране на тежки деформации на крайниците и възстановяване на тяхното функциониране. За тази цел може да се извърши остеоклазия, остеотомия с помощта на фиксатори и метална остеосинтеза; понякога се използват дистракционно-компресиращи устройства. Операцията може да се извърши само след като детето навърши 5 години. Показания за операция са тежки деформации на крайниците, които затрудняват избора на ортопедични устройства, които да помогнат на пациента да се движи. При подготовката за операция на пациентите се прилага описаното по-горе консервативно лечение.
Терапевтичните упражнения се провеждат внимателно за такива пациенти. Физическите упражнения, изпълнявани в топла вода, станаха широко разпространени.
Масажът се основава на техники на поглаждане и разтриване.
Преди операцията на пациентите се предписват терапевтични упражнения и масаж за укрепване на мускулите, в следоперативния период са показани изометрични упражнения.
Основната цел на лечението на остеогенезис имперфекта е да се постигне способността на пациента да стои в ортопедични устройства и да развие умения за движение в тях.
По този начин остеогенезис имперфекта е сериозно наследствено заболяване, което изисква прилагането на обширна физиотерапевтична програма, постоянни хирургични интервенции при скелетни деформации и фрактури, както и специално обучение и психологическа подкрепа за пациента и неговите близки.
